


The Module 3 of a Common Technical Document (CTD) also known as the Quality (CMC) section of a regulatory dossier provides detailed data on the composition, manufacturing process, quality controls, and stability of the Drug Substance (API) and Drug Product (finished product).
For regulatory teams, the primary challenge is not the 'writing' of the CMC section itself, but rather compiling the right data from a large set of source input files and then keeping every compiled statement or data traceable back to its origin. These 'source files' typically include a comprehensive set of technical, analytical, and manufacturing documentation to prove that a drug substance (API) and drug product (finished dosage form) are manufactured consistently, safely, and to high-quality standards.
The challenge: the document burden is real
On average, an NDA contains well over 100,000 pages of information (including summaries of information the sponsor has gathered). Even ANDAs, which are typically smaller than NDAs still involve compiling and tracing 1000s of pages of source input documents, especially across the quality/CMC portion of the submission.
At the same time, many organizations generate large numbers of CMC reports continuously across development and lifecycle management. Dassault Systèmes (BIOVIA) describes it as not unusual for large pharma/biotech companies to generate hundreds of reports each year, with reports often approaching 100 pages and pulling data from multiple sources.
This is the reality regulatory teams deal with: a large number of input documents that need to be compiled into a structured dossier, kept consistent, and defended under review.
Why it matters: quality and manufacturing issues block approvals
In July 2025, FDA published more than 200 Complete Response Letters (CRLs) issued between 2020 and 2024, creating a rare window into what derails first-cycle approvals. (Source: FDA) A separate analysis of 202 CRLs reported that 150 (74%) involved quality and manufacturing issues, including facility/process concerns. (Source: PharmaManufacturing)
Compiling and verifying sources is time-consuming and can slow down even the highest-performing teams. Every key statement in the CMC section eventually has to be backed up with specifics: which source input document supports it, which version was used, and whether the same claim stays consistent everywhere it appears across the module.
Where time actually goes
If you strip the work down to the steps that consume time, the slow parts are usually consistent.
- Intake and triage of source input documents: Teams have to scour through multiple source input files (e.g., stability reports, manufacturing documents, batch analysis, etc.) to find the specific data and values to copy into the dossier.
- Manual compilation into the CTD structure: Even with a clear outline, compilation is repetitive when it depends on manual extraction and re-entry. The same underlying information often has to appear in multiple sections, which increases the chance that edits drift over time.
- Review cycles get spent on document hunting: When traceability is not established during compilation, it gets rebuilt later during internal review, response preparation, or submission readiness checks. Reviewers waste significant time trying to match a statement or a data point in the dossier to the original supporting document.
- Reconciliation across parallel edits: As contributions come in from different owners, inconsistencies appear. Even small differences can trigger review chains: language that no longer matches the most recent source, a value updated in one place but not another, or a conclusion that is no longer aligned with the referenced evidence.
So when teams say “CMC drafting takes too long,” what they often mean is “it takes too long to prove that everything lines up.”
What “solving it” looks like in practice
At SyncIQ, we believe the solution is to eliminate manual copy-pasting. We treat CMC writing as a dynamic output where every data point remains electronically linked to its source file, rather than a static document where data is pasted and disconnected.
In practice, the solution is straightforward:
Generate drafts from linked evidence, then apply expertise.
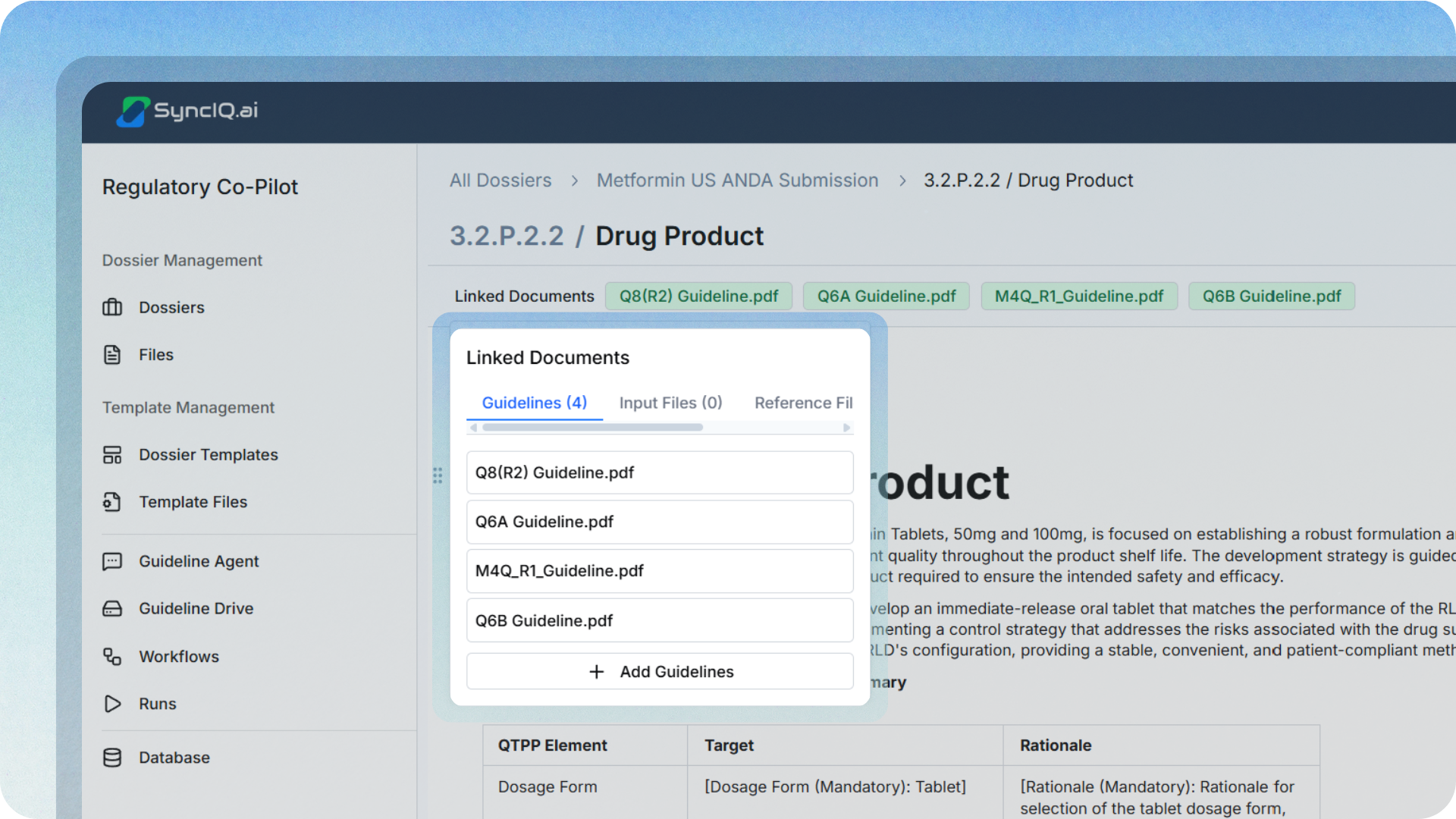
Generate drafts based on the linked source files, then allow subject matter experts to refine the content with the right guideline expectations already “in the workflow.” The goal is not to remove human judgment. It is to remove the manual extraction and reconciliation so experts can focus on interpretation, risk framing, and decision quality.
Move from folders to a structured data room.
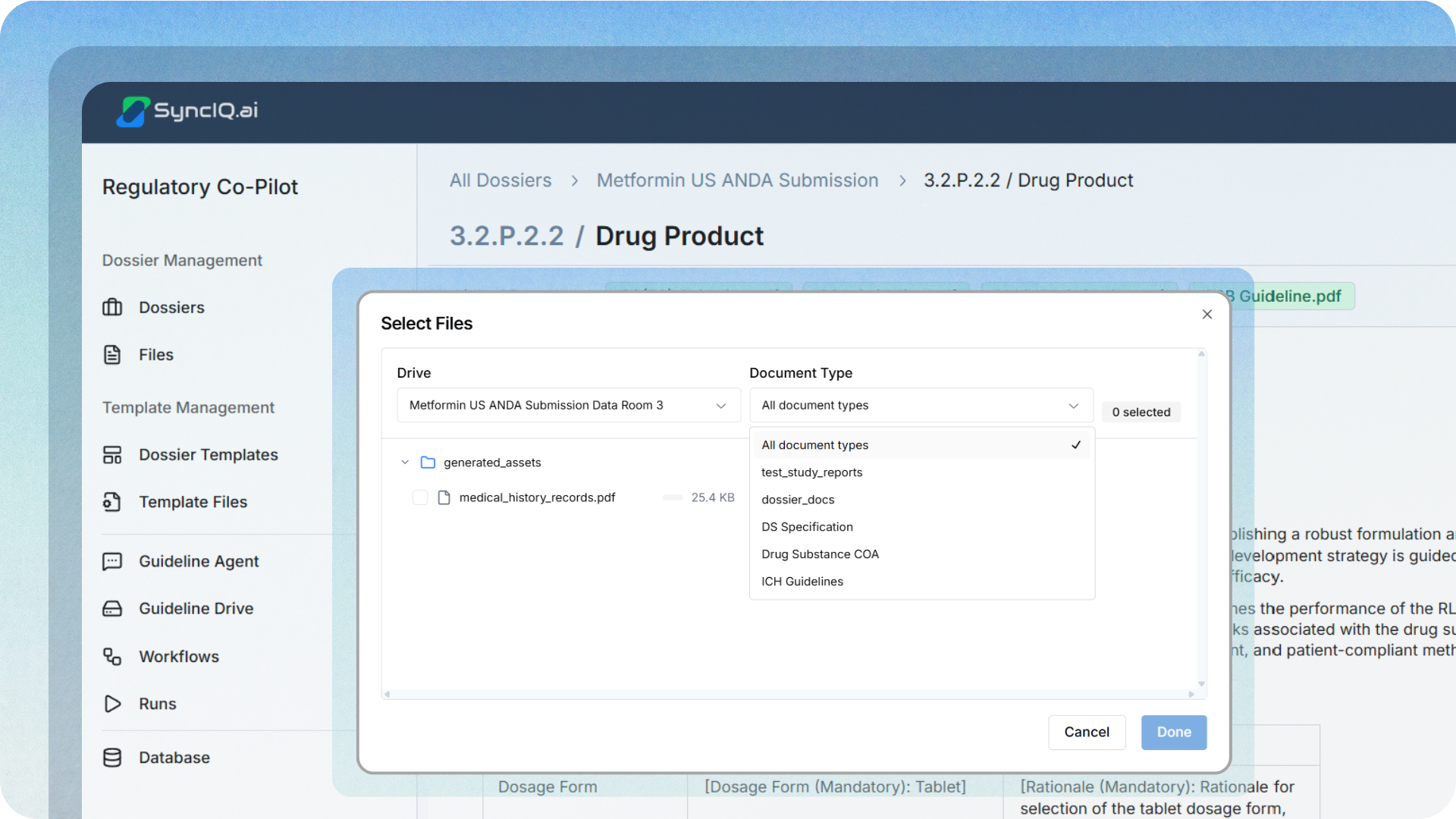
The raw source input files need to be organized so compilation is deterministic. Specs, methods, validations, stability outputs, and manufacturing narratives should be classified and easy to find by their content type, rather than relying on a user's memory of a folder structure.
Make traceability the default.
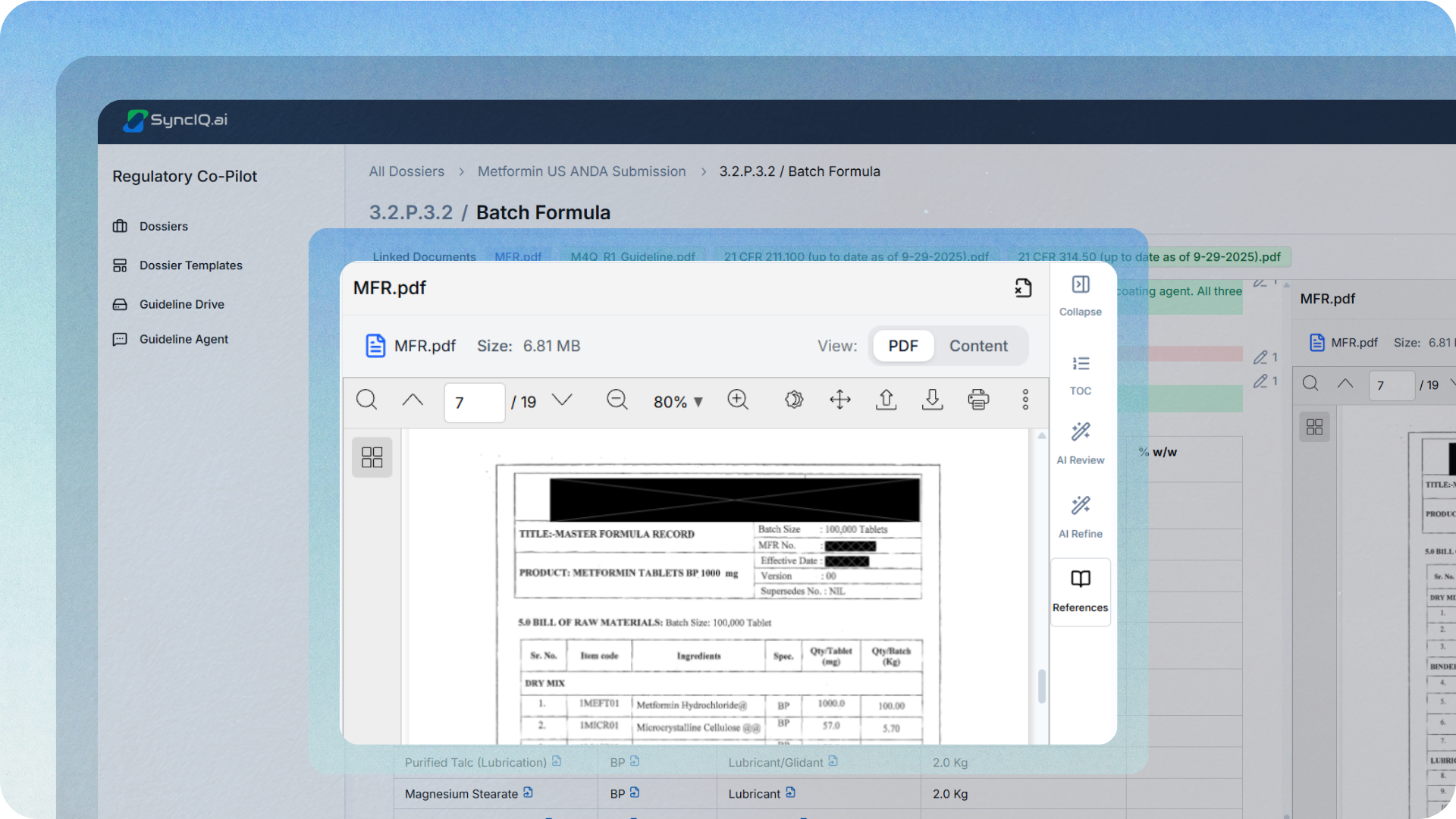
Key claims should be linked to specific sources and versions. Not “this came from the stability report,” but “this conclusion is supported by this table in this version of the report.” That is how authoring becomes reviewable and defensible.
How SyncIQ fits into this
This is the direction we have taken with our Regulatory CoPilot: focus on making compilation more systematic and traceability easier to maintain. The goal is not to “replace writers.” It is to reduce the two costs that dominate CMC authoring: time lost finding the right evidence and time lost proving it.
Try a quick self-test: pick five high-impact statements in your current workflow and ask, “Can the team prove the source and version behind each statement in under 10 minutes, and can they identify every dependent section that must update if the source changes?” If the answer is “not consistently,” then this is not a writing problem. It is a compilation and traceability problem.

